L'approccio matematico può prevedere la struttura cristallina in ore invece di mesi.
14 novembre 2024
Questo articolo è stato revisionato secondo il processo editoriale e le politiche di Science X. Gli editor hanno evidenziato i seguenti attributi garantendo la credibilità del contenuto:
- verifica dei fatti
- pubblicazione sottoposta a revisione paritaria
- fonte affidabile
corretto da Rachel Harrison, Università di New York
I ricercatori presso l'Università di New York hanno elaborato un approccio matematico per prevedere le strutture dei cristalli: un passo critico nello sviluppo di molti medicinali e dispositivi elettronici, in poche ore utilizzando solo un laptop, un processo che in precedenza richiedeva settimane o mesi a un supercomputer. Il loro nuovo framework è pubblicato sulla rivista Nature Communications.
I cristalli molecolari organici sono una classe importante di materiali in molte industrie, dalla farmaceutica e l'agricoltura all'elettronica e agli esplosivi. I cristalli sono i mattoni che costituiscono molti farmaci da banco e da prescrizione, insetticidi per combattere le zanzare, esplosivi come TNT, semiconduttori e tecnologie emettitrici di luce utilizzate negli schermi televisivi e nei cellulari.
Nonostante l'ubiquità dei cristalli molecolari in molti prodotti di uso quotidiano, la capacità di prevederne le strutture tridimensionali rimane una sfida, specialmente se un composto può cristallizzare in forme multiple. In un esempio drammatico della necessità di prevedere le strutture cristalline, gli scienziati alla fine degli anni '90 scoprirono che le capsule del farmaco anti-HIV ritonavir si trasformarono successivamente dalla forma cristallina conosciuta in una forma sconosciuta ma più stabile. Questo cambiamento nella struttura cristallina rendeva il farmaco inefficace e lo costrinse a uscire dal mercato fino a quando non fu creata una nuova formulazione.
La maggior parte degli approcci attuali per la previsione delle strutture cristalline utilizzano metodi basati sulla fisica che hanno limitazioni, tra cui l'introduzione di bias ed errori o la previsione di troppe forme cristalline rispetto a quelle che si verificano effettivamente negli esperimenti. Inoltre, tali metodi richiedono una potenza di calcolo significativa e possono richiedere settimane o mesi, a seconda della complessità delle molecole costituenti.
'Questi approcci basati sulla fisica, costosi e che richiedono molto tempo, producono previsioni che sono accurate solo quanto la fisica che vi metti dentro, ecco perché c'è stata una spinta verso metodi computazionali che possano affrontare questa lacuna', ha detto Mark Tuckerman, professore di chimica e matematica presso la NYU e autore senior dello studio.
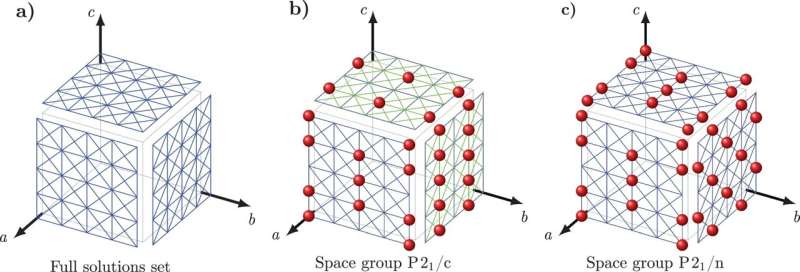
Per superare questa sfida, Tuckerman e Nikolaos Galanakis, un ricercatore postdottorato presso la NYU, hanno sviluppato un nuovo approccio matematico che hanno chiamato 'Crystal Math' per prevedere le strutture cristalline basate esclusivamente su regole matematiche che regolano come le molecole si impacchettano nei cristalli e su alcuni semplici descrittori fisici dell'ambiente del cristallo.
Crystal Math risolve quindi 13 parametri di base relativi all'organizzazione delle molecole nel cristallo, inclusa la posizione e l'orientamento molecolare e la geometria dei blocchi di base del cristallo, oltre ad altri fattori geometrici che definiscono la forma di ogni molecola nel cristallo.
Tuckerman e Galanakis hanno verificato le regole di Crystal Math utilizzando il Cambridge Crystal Data Centre, un database di centinaia di migliaia di strutture cristalline molecolari organiche conosciute. In particolare, i ricercatori hanno testato se le loro regole matematiche ipotizzate venivano rispettate dalle strutture nel database, che li ha guidati verso principi che le strutture conosciute seguiranno molto probabilmente.
Successivamente, hanno inserito questi principi in un insieme di equazioni il cui risultato può ora essere utilizzato per prevedere le strutture cristalline molecolari non presenti nel database. Farmaci comuni come l'aspirina e il paracetamolo, le cui strutture sono già conosciute, sono stati utilizzati come casi di test semplici.
Successivamente, utilizzando le equazioni di Crystal Math, i ricercatori hanno applicato la procedura a cristalli molecolari più complessi, inclusi molecole altamente flessibili, le cui strutture non sono presenti nel database, ottenendo previsioni di strutture che corrispondevano a quelle generate negli esperimenti con alta accuratezza.
'Le nostre equazioni sembrano, finora, darci solo strutture cristalline sperimentabilmente realizzabili, il che affronta il problema dei metodi basati sulla fisica che tendono a 'sovrastimare' il numero di possibili strutture, il che significa che alcune delle strutture previste potrebbero non essere mai trovate sperimentalmente', ha detto Tuckerman.
È importante sottolineare che le soluzioni possono essere ottenute in poche ore su un normale laptop, anziché richiedere i lunghi tempi e i computer ad alta performance necessari per i metodi basati sulla fisica.
'Il tempo per ottenere una soluzione non è più di settimane o mesi: possiamo ottenere una soluzione durante la notte perché risolvere le equazioni è relativamente veloce', ha aggiunto Tuckerman.
Crystal Math rappresenta il culmine di sette anni di lavoro di Tuckerman e Galanakis per ideare una soluzione matematica a questo grande problema. Tuckerman è stato particolarmente ispirato da un articolo del 1967 del matematico e cristallografo svizzero Johann Jakob Burckhardt, che suggeriva che fosse possibile utilizzare la matematica per prevedere le strutture cristalline, ma non ha presentato una soluzione propria.
Oltre 55 anni dopo, il protocollo basato sulla matematica di Tuckerman e Galanakis ha suscitato interesse nell'industria farmaceutica e promette di indagare su composti ancora da scoprire e prevedere le loro strutture cristalline.
'La capacità di sviluppare nuovi prodotti dipende dal sapere se i composti che li costituiscono si cristallizzeranno, quante forme cristalline sono possibili e la stabilità di queste forme diverse,' ha detto Tuckerman. 'Con il nostro approccio matematico, è possibile testare la capacità di molti composti di cristallizzarsi e determinare se queste strutture sono adatte per un'eventuale commercializzazione sul mercato.'
Maggiori informazioni: Nikolaos Galanakis et al, Rapida previsione delle strutture cristalline molecolari utilizzando semplici descrittori topologici e fisici, Nature Communications (2024). DOI: 10.1038/s41467-024-53596-5
Informazioni sulla rivista: Nature Communications
Fornito da New York University



