Matematisk metod kan förutsäga kristallstruktur på några timmar istället för månader
14 november 2024
Den här artikeln har granskats enligt Science X:s redaktionella process och riktlinjer. Redaktörerna har framhävt följande egenskaper samtidigt som de säkerställer innehållets trovärdighet:
- faktagranskad
- vänreferentgranskad publicering
- pålitlig källa
- korrekturläst
av Rachel Harrison, New York University
Forskare vid New York University har utvecklat ett matematiskt tillvägagångssätt för att förutsäga strukturerna av kristaller - ett avgörande steg för att utveckla många läkemedel och elektroniska enheter - på några timmar med endast en bärbar dator, en process som tidigare tog en superdator veckor eller månader. Deras nyskapande ramverk publiceras i tidskriften Nature Communications.
Organiska molekylära kristaller är en viktig materialklass inom många branscher, från läkemedel och jordbruk till elektronik och sprängmedel. Kristaller är byggstenarna som utgör många receptfria och receptbelagda läkemedel, insekticider för att bekämpa myggor, sprängämnen som TNT, halvledare samt ljusemitterande teknologier som används i televisionsskärmar och mobiltelefoner.
Trots förekomsten av molekylära kristaller i många vardagliga produkter är förmågan att förutsäga deras tredimensionella strukturer fortfarande en utmaning, särskilt om en förening kan kristalliseras i flera former. I ett dramatiskt exempel på nödvändigheten att förutsäga kristallstrukturer, upptäckte forskare i slutet av 1990-talet att kapslar av HIV-läkemedlet ritonavir senare omvandlades från den kända kristallformen till en okänd men mer stabil form. Denna förändring i kristallstrukturen gjorde läkemedlet ineffektivt och tvingade det ur marknaden tills en ny formulering skapades.
De flesta nuvarande tillvägagångssätt för att förutsäga kristallstrukturer använder fysikbaserade metoder som har begränsningar, inklusive att införa partiskhet och fel eller förutsäga för många kristallformer än de som faktiskt uppstår i experiment. Dessutom kräver metoderna betydande databehandlingskraft och kan ta veckor till månader, beroende på komplexiteten hos de ingående molekylerna.
”Dessa fysikbaserade tillvägagångssätt - som är kostsamma och tidskrävande - ger förutsägelser som bara är lika exakta som fysiken du matar in i dem, vilket är varför det har funnits en strävan mot beräkningsmetoder som kan ta itu med detta bristfällighet,” sa Mark Tuckerman, professor i kemi och matematik vid NYU och den äldre författaren till studien.
För att övervinna denna utmaning, utvecklade Tuckerman och Nikolaos Galanakis, en postdoktorand vid NYU, ett nytt matematiskt tillvägagångssätt som de har döpt till 'Crystal Math' för att förutsäga kristallstrukturer baserat på rent matematiska regler som styr hur molekyler packas in i kristaller och några få enkla fysikaliska beskrivare av kristallens miljö.
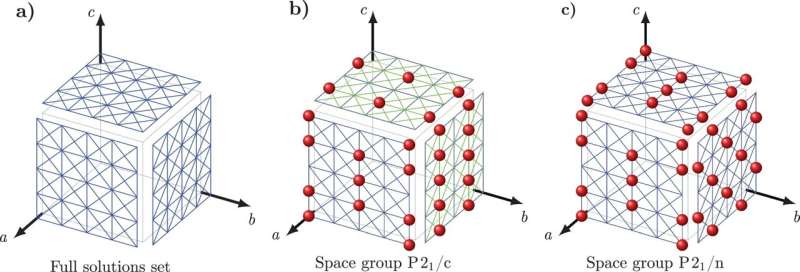
Crystal Math löser sedan 13 grundläggande parametrar relaterade till arrangemanget av molekyler i kristallen - inklusive molekylär plats och orientering och geometrin hos kristallens grundläggande byggstenar - plus andra geometriska faktorer som definierar formen på varje molekyl i kristallen.
Tuckerman och Galanakis verifierade Crystal Maths regler genom att använda Cambridge Crystal Data Center, en databas med hundratusentals kända organiska molekylära kristallstrukturer. Specifikt testade forskarna om deras hypotetiserade matematiska regler följdes av strukturerna i databasen, vilket ledde dem till principer som kända strukturer hade mycket sannolikhet att följa.
Sedan byggde de in dessa principer i en uppsättning ekvationer vars lösningar nu kan användas för att förutsäga molekylära kristallstrukturer som inte finns i databasen. Vanliga läkemedel som aspirin och paracetamol, vars strukturer redan är kända, användes som enkla testfall.
Nästa steg var att använda Crystal Maths ekvationer för att tillämpa deras procedur på mer komplexa molekylära kristaller, inklusive mycket flexibla molekyler, vars strukturer inte finns i databasen, och erhöll strukturförutsägelser som stämde överens med dem som genererats i experiment med hög noggrannhet.
”Våra ekvationer ger tydligen än så länge endast kristallstrukturer som kan realiseras experimentellt, vilket tacklar problemet med fysikbaserade metoder som tenderar att 'överförutsäga' antalet möjliga strukturer, vilket innebär att några av de förutsagda strukturerna aldrig kan hittas experimentellt,” sa Tuckerman.
Viktigt är att lösningarna kan uppnås på bara några få timmar på en vanlig bärbar dator, istället för att kräva de långa tidsramarna och högpresterande datorer som krävs av fysikbaserade metoder.
”Tid till lösningen är inte längre veckor till månader - vi kan få en lösning över natten eftersom det är relativt snabbt att lösa ekvationerna,” tillade Tuckerman.
Crystal Math representerar kulmen av sju års arbete av Tuckerman och Galanakis för att hitta en matematisk lösning på denna stora utmaning. Tuckerman blev särskilt inspirerad av en artikel från 1967 av schweiziska matematikern och kristallografen Johann Jakob Burckhardt, som föreslog att det skulle vara möjligt att använda matematik för att förutsäga kristallstrukturer, men inte presenterade en lösning själv.
Mer än 55 år senare har Tuckerman och Galanakis matematikbaserade protokoll väckt intresse från läkemedelsindustrin och lovar att undersöka ännu ej upptäckta föreningar och förutse deras kristallstrukturer.
"Förmågan att utveckla nya produkter beror på att veta om de föreningar som utgör dem kommer att kristallisera, hur många kristallformer som är möjliga, och stabiliteten hos dessa olika former", sa Tuckerman. "Med vårt matematiska tillvägagångssätt är det möjligt att testa förmågan hos många föreningar att kristallisera och avgöra om dessa strukturer är lämpliga för slutgiltig användning på marknaden."
Mer information: Nikolaos Galanakis et al, Rapid prediction of molecular crystal structures using simple topological and physical descriptors, Nature Communications (2024). DOI: 10.1038/s41467-024-53596-5
Tidskriftsinformation: Nature Communications
Med tillstånd av New York University



