Podejście matematyczne może przewidzieć strukturę kryształu w ciągu kilku godzin zamiast miesięcy
14 listopada 2024
Ten artykuł został przejrzany zgodnie z procesem redakcyjnym i politykami Science X. Redaktorzy podkreślili następujące cechy, zapewniając wiarygodność treści:
- zweryfikowane pod względem faktów
- publikacja z recenzjami innych naukowców
- zaufane źródło
- skorygowane
przez Rachel Harrison z Uniwersytetu w Nowym Jorku
Naukowcy z Uniwersytetu w Nowym Jorku opracowali podejście matematyczne do przewidywania struktur kryształów—kluczowego kroku w rozwoju wielu leków i urządzeń elektronicznych—w zaledwie kilka godzin, korzystając tylko z laptopa, proces ten wcześniej zajmował superkomputerom tygodnie lub miesiące. Ich nowatorska metoda została opublikowana w czasopiśmie Nature Communications.
Organiczne kryształy molekularne są ważną klasą materiałów w wielu branżach, od farmaceutycznej i rolniczej do elektronicznej i wybuchowej. Kryształy są budulcem wielu leków, insektycydów do walki z komarami, materiałów wybuchowych takich jak TNT, półprzewodników oraz technologii emitujących światło, używanych w ekranach telewizyjnych i telefonach komórkowych.
Mimo powszechności kryształów molekularnych w wielu codziennych produktach, przewidywanie ich struktur trójwymiarowych pozostaje wyzwaniem, szczególnie jeśli związek może krystalizować w kilku formach. W jednym dramatycznym przykładzie konieczności przewidywania struktur kryształowych, w latach 90. naukowcy odkryli, że kapsułki leku na HIV, ritonavir, później przekszałciły się z znanej formy kryształu w nieznaną, ale bardziej stabilną formę. Zmiana w strukturze kryształu sprawiła, że lek stał się nieskuteczny i został wycofany z rynku, dopóki nie stworzono nowej formuły.
Obecne metody przewidywania struktur kryształowych wykorzystując fizykę mają pewne ograniczenia, np. wprowadzanie błędów, przewidywanie zbyt wielu form kryształów niż te, które faktycznie pojawiają się w eksperymentach. Ponadto, metody te wymagają dużej mocy obliczeniowej i mogą trwać tygodnie lub miesiące, w zależności od złożoności składników molekularnych.
'Te podejścia oparte na fizyce—które są kosztowne i czasochłonne—oprogramiają przewidywania tylko na podstawie fizyki, którą do nich wprowadzasz, dlatego pojawiła się potrzeba metod obliczeniowych, które mogą przezwyciężyć tę wadę,' powiedział Mark Tuckerman, profesor chemii i matematyki na NYU i główny autor badania.
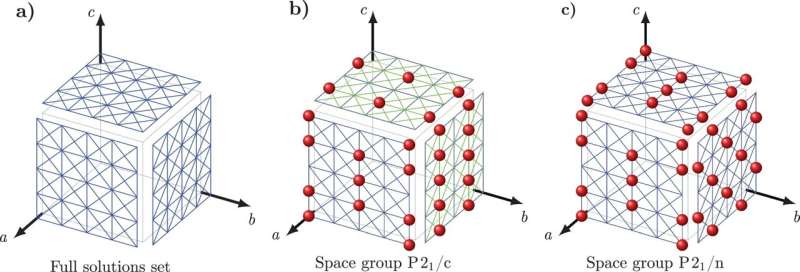
Aby pokonać to wyzwanie, Tuckerman i Nikolaos Galanakis, badacz podyplomowy na NYU, opracowali nowe podejście matematyczne nazwane 'Matematyka Kryształów' do przewidywania struktur kryształowych oparte na czysto matematycznych zasadach rządzących tym, w jaki sposób molekuły pakują się w kryształy oraz na kilku prostych deskryptorach fizycznych otoczenia kryształu.
Matematyka Kryształów oblicza 13 podstawowych parametrów związanych z ułożeniem molekuł w krysztale—w tym położenie i orientacja molekuł oraz geometria podstawowych jednostek budulcowych kryształu—i inne czynniki geometryczne, które określają kształt każdej molekuły w krysztale.
Tuckerman i Galanakis zweryfikowali zasady Matematyki Kryształów, korzystając z Cambridge Crystal Data Centre, bazy danych zawierającej setki tysięcy znanych struktur organicznych kryształów molekularnych. Konkretnie, naukowcy sprawdzili, czy hipotetyczne zasady matematyczne były przestrzegane przez struktury w bazie danych, co zaprowadziło ich do zasad, które znane struktury były bardzo prawdopodobne, że będą przestrzegać.
Następnie zbudowali te zasady w zestaw równań, których rozwiązania mogą teraz być używane do przewidywania struktur kryształowych molekuł nieznalezionych w bazie danych. Powszechne leki takie jak aspiryna i paracetamol, których struktury są już znane, były używane jako proste przypadki testowe.
Następnie, korzystając z równań Matematyki Kryształów, naukowcy zastosowali swoją metodę do bardziej złożonych kryształów molekularnych, w tym o bardzo dużym stopniu elastyczności molekuł, których struktury nie ma w bazie danych, i uzyskali przewidywania struktury, które zgadzały się z wynikami eksperymentów z dużą dokładnością.
'Nasze równania, jak na razie, wydają się dawać nam tylko eksperymentalnie realizowalne struktury kryształów, co rozwiązuje problem metod opartych na fizyce, które mają tendencję do 'przewidywania zbyt wielu' możliwych struktur, co oznacza, że niektóre z przewidywanych struktur mogą nigdy nie zostać znalezione eksperymentalnie,' powiedział Tuckerman.
Co ważne, rozwiązania mogą być osiągnięte w zaledwie kilka godzin na standardowym laptopie, zamiast wymagać długich skal czasowych i komputerów o wysokiej wydajności, jak ma to miejsce w przypadku metod opartych na fizyce.
'Czas uzyskania rozwiązania to już nie tygodnie czy miesiące—możemy uzyskać rozwiązanie w ciągu nocy, ponieważ rozwiązywanie równań jest stosunkowo szybkie,' dodał Tuckerman.
Kryształowa Matematyka stanowi kulminację siedmiu lat pracy Tuckermana i Galanakisa nad opracowaniem matematycznego rozwiązania tego wielkiego problemu. Tuckerman szczególnie inspirował się pracą szwajcarskiego matematyka i krystalografa Johanna Jakoba Burckhardta z 1967 roku, który sugerował, że możliwe powinno być wykorzystanie matematyki do przewidywania struktur krystalicznych, ale sam nie zaproponował rozwiązania.
Ponad 55 lat później protokół oparty na matematyce Tuckermana i Galanakisa wzbudza zainteresowanie w przemyśle farmaceutycznym i obiecuje badanie nieodkrytych jeszcze związków i przewidywanie ich struktur krystalicznych.
"Możliwość opracowania nowych produktów zależy od wiedzy, czy związki, które je tworzą, będą ulegały krystalizacji, ile form krystalicznych jest możliwych, oraz od stabilności tych różnych form" - powiedział Tuckerman. "Dzięki naszemu podejściu matematycznemu możliwe jest przetestowanie zdolności wielu związków do krystalizacji i ustalenie, czy te struktury są odpowiednie do ostatecznego wdrożenia na rynek".
Więcej informacji: Nikolaos Galanakis et al, Rapid prediction of molecular crystal structures using simple topological and physical descriptors, Nature Communications (2024). DOI: 10.1038/s41467-024-53596-5
Informacje o czasopiśmie: Nature Communications
Dostarczone przez New York University



