Wiskundige benadering kan de kristalstructuur voorspellen in uren in plaats van maanden
14 november 2024
Dit artikel is beoordeeld volgens het redactionele proces en de beleidsregels van Science X. Redacteuren hebben de volgende kenmerken benadrukt terwijl ze de geloofwaardigheid van de inhoud waarborgen:
- feiten gecheckt
- door vakgenoten beoordeeld tijdschrift
- vertrouwde bron
- nagekeken
door Rachel Harrison, New York University
Onderzoekers aan de New York University hebben een wiskundige benadering bedacht om de structuren van kristallen te voorspellen - een cruciale stap bij de ontwikkeling van veel medicijnen en elektronische apparaten - in slechts enkele uren met behulp van alleen een laptop, een proces dat voorheen weken of maanden kostte met een supercomputer. Hun nieuwe raamwerk is gepubliceerd in het tijdschrift Nature Communications.
Organische moleculaire kristallen zijn een belangrijke klasse materialen in veel industrieën, van farmaceutica en landbouw tot elektronica en explosieven. Kristallen zijn de bouwstenen die veel vrij verkrijgbare en voorgeschreven medicijnen, insecticiden tegen muggen, explosieven zoals TNT, halfgeleiders, en lichtgevende technologieën die worden gebruikt in televisieschermen en mobiele telefoons vormen.
Ondanks het alomtegenwoordige van moleculaire kristallen in veel alledaagse producten, blijft het een uitdaging om hun driedimensionale structuren te voorspellen, vooral als een verbinding in meerdere vormen kan kristalliseren. In een dramatisch voorbeeld van de noodzaak om kristalstructuren te voorspellen, ontdekten wetenschappers eind jaren negentig dat capsules van het HIV-medicijn ritonavir later veranderden van de bekende kristalvorm naar een onbekende maar stabielere vorm. Deze verandering in kristalstructuur maakte het medicijn ineffectief en dwong het van de markt tot er een nieuwe formulering werd gemaakt.
De meeste huidige benaderingen voor het voorspellen van kristalstructuren gebruiken op de natuurkunde gebaseerde methoden die beperkingen hebben, waaronder het introduceren van vooringenomenheid en fouten of het voorspellen van te veel kristalvormen dan er daadwerkelijk in experimenten voorkomen. Bovendien vereisen de methoden aanzienlijke rekencapaciteit en kunnen weken tot maanden duren, afhankelijk van de complexiteit van de samenstellende moleculen.
'Deze kosten- en tijdsintensieve op natuurkunde gebaseerde benaderingen produceren voorspellingen die slechts zo accuraat zijn als de natuurkunde die je erin stopt, daarom is er een verschuiving geweest naar rekenmethoden die dit tekort kunnen aanpakken,' zei Mark Tuckerman, professor in de chemie en wiskunde aan de NYU en senior auteur van het onderzoek.
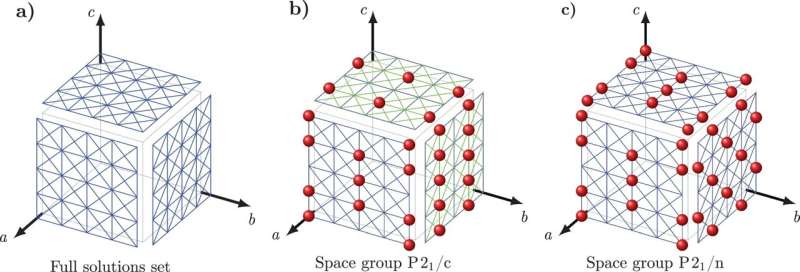
Om deze uitdaging aan te gaan, ontwikkelden Tuckerman en Nikolaos Galanakis, een postdoctoraal onderzoeker van de NYU, een nieuwe wiskundige benadering die ze 'Crystal Math' hebben genoemd om kristalstructuren te voorspellen op basis van louter wiskundige regels die bepalen hoe moleculen zich in kristallen verpakken en enkele eenvoudige fysieke kenmerken van de omgeving van het kristal.
Crystal Math berekent vervolgens 13 basisparameters met betrekking tot de rangschikking van moleculen in het kristal - inclusief de locatie en oriëntatie van moleculen en de geometrie van de basis bouwstenen van het kristal - plus andere geometrische factoren die de vorm van elk molecuul in het kristal bepalen.
Tuckerman en Galanakis hebben de regels van Crystal Math geverifieerd met behulp van het Cambridge Crystal Data Centre, een database van honderdduizenden bekende organische moleculaire kristalstructuren. De onderzoekers hebben specifiek getest of hun veronderstelde wiskundige regels worden nageleefd door de structuren in de database, wat hen leidde naar principes die bekende structuren zeer waarschijnlijk volgen.
Vervolgens hebben ze deze principes omgezet in een reeks vergelijkingen waarvan de oplossingen nu kunnen worden gebruikt om moleculaire kristalstructuren te voorspellen die niet in de database voorkomen. Gangbare farmaceutica zoals aspirine en paracetamol, waarvan de structuren al bekend zijn, werden gebruikt als eenvoudige testgevallen.
Vervolgens pasten de onderzoekers met behulp van de vergelijkingen van Crystal Math hun procedure toe op complexere moleculaire kristallen, waaronder zeer flexibele moleculen, waarvan de structuren niet in de database voorkomen en verkregen structuurvoorspellingen die overeenkwamen met die welke werden gegenereerd in experimenten met hoge nauwkeurigheid.
'Tot nu toe lijken onze vergelijkingen uitsluitend experimenteel realiseerbare kristalstructuren te geven, wat het probleem van op natuurkunde gebaseerde methoden die de neiging hebben het aantal mogelijke structuren te 'overvoorspellen' en dan leiden tot voorspellingen die experimenteel niet worden gevonden, aanpakt,' zei Tuckerman.
Belangrijk is dat de oplossingen in slechts enkele uren op een standaard laptop kunnen worden bereikt, in plaats van de lange tijdschalen en hoogwaardige computers die nodig zijn voor natuurkundige methoden.
'De oplossing is niet langer weken tot maanden — we kunnen 's nachts een oplossing krijgen omdat het oplossen van de vergelijkingen relatief snel is,' voegde Tuckerman toe.
Crystal Math vertegenwoordigt het resultaat van zeven jaar werk van Tuckerman en Galanakis om een wiskundige oplossing te bedenken voor dit groot uitdaging probleem. Tuckerman werd in het bijzonder geïnspireerd door een paper uit 1967 van de Zwitserse wiskundige en kristallograaf Johann Jakob Burckhardt, die suggereerde dat het mogelijk zou moeten zijn om wiskunde te gebruiken om kristalstructuren te voorspellen, maar zelf geen oplossing naar voren bracht.
Meer dan 55 jaar later heeft het op wiskunde gebaseerde protocol van Tuckerman en Galanakis interesse gewekt in de farmaceutische industrie en belooft het onderzoek naar nog niet ontdekte verbindingen mogelijk te maken en hun kristalstructuren te voorspellen.
'Het vermogen om nieuwe producten te ontwikkelen hangt in hoge mate af van het weten of de verbindingen die ze vormen zullen kristalliseren, hoeveel kristalvormen er mogelijk zijn en de stabiliteit van deze verschillende vormen,' aldus Tuckerman. 'Met onze wiskundige aanpak is het mogelijk om de kristallisatiecapaciteit van vele verbindingen te testen en te bepalen of deze structuren geschikt zijn voor uiteindelijke toepassing op de markt.'
Meer informatie: Nikolaos Galanakis et al, Snelle voorspelling van moleculaire kristalstructuren met eenvoudige topologische en fysische beschrijvingen, Nature Communications (2024). DOI: 10.1038/s41467-024-53596-5
Tijdschriftinformatie: Nature Communications
Verstrekt door New York University



